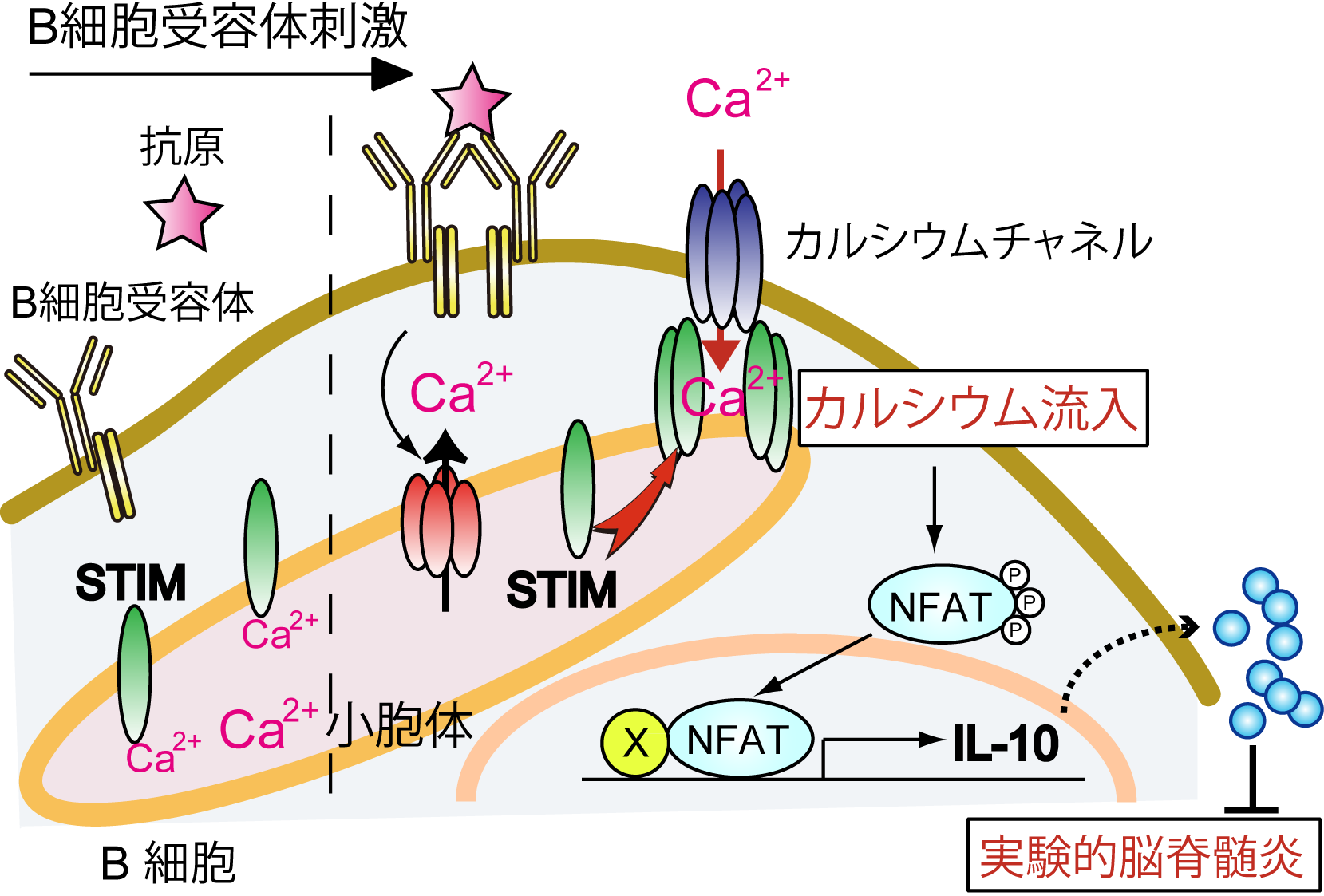
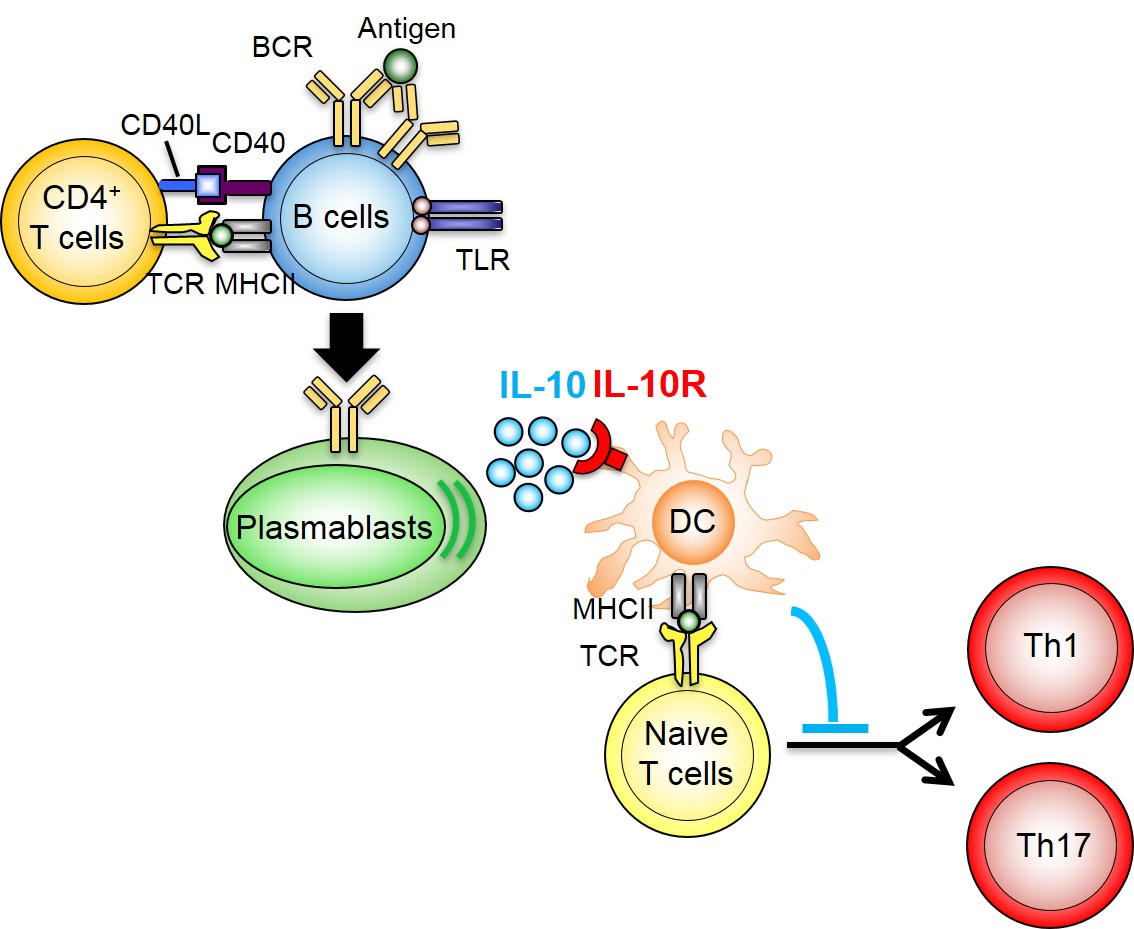
図1 抗原刺激時の脾臓における記憶B細胞の分化
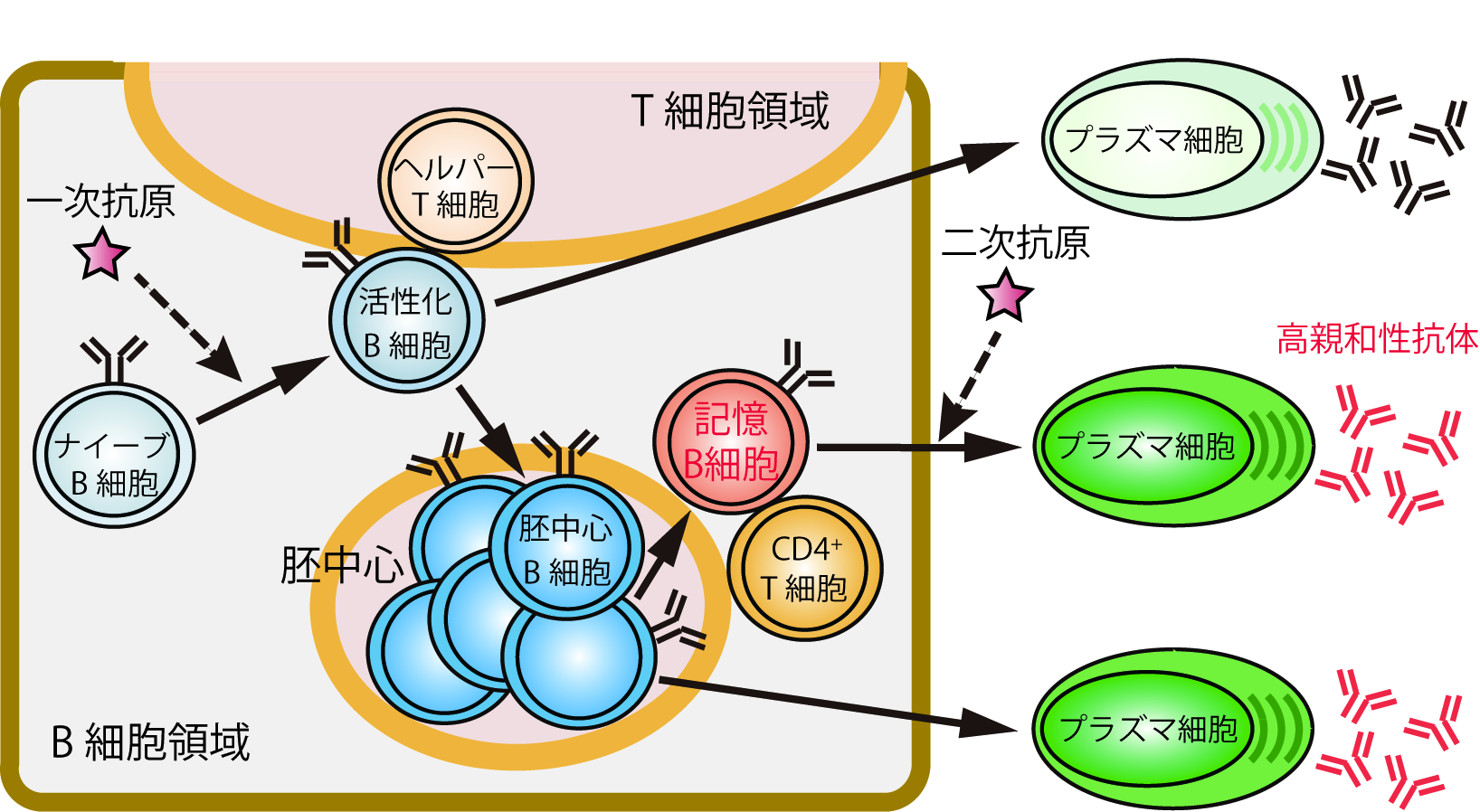
ナイーブB細胞は抗原(一次抗原)に感作されて活性化されると、ヘルパーT細胞からの刺激を受けてプラズマ細胞や胚中心B細胞へと分化する。胚中心B細胞の一部はプラズマ細胞へ分化するが、残りの一部は記憶B細胞へと分化して胚中心の近傍で長期間維持される。この記憶B細胞は、再度、同じ抗原(二次抗原)による刺激を受けると、CD4陽性のT細胞(CD4+ T細胞)のヘルプを受けてプラズマ細胞へと分化し、抗原に対して高親和性の抗体を産生する。
現在、我々の研究グループでは、記憶B細胞による感染防御システムを人為的に構築することを目指して、以下の疑問点を明らかにしようとしています。
- 記憶B細胞がどのように産生され、長期間維持されているのか?
- 記憶B細胞がどのようなメカニズムで迅速で強い抗体産生能を獲得しているのか?
- CD4陽性のT細胞がどのようにして記憶B細胞による抗体産生を誘導しているか?